Deze week worden de Nobelprijzen weer uitgereikt en zojuist is die voor Scheikunde bekend gemaakt. Voordat ik het onderwerp van dit jaar zal uitleggen, kan ik alvast verklappen: als de Nobelprijs aan meer dan 3 mensen tegelijk uitgereikt mocht worden, zaten er zéker een aantal Nederlanders tussen. Desalniettemin is het onderwerp ontzettend gaaf en ik zal hieronder in lekentaal uit de doeken doen, waarom deze drie mannen wonnen.

Elk onderzoek wordt gedaan met Een Grote Vraag. Bij die Grote Vraag horen subvragen, en die worden op diens beurt weer onderverdeeld in nog kleinere subvragen. Verreweg de meeste onderzoekers werken aan kleine subsubsubvragen — zogenaamd ‘fundamenteel onderzoek’ — waarbij de toepassing ver weg is. De Grote Vraag die hier wordt gesteld is: kunnen we perfecte medicijnen berekenen met een computer, in plaats van ze te testen op dieren. Dit is een variatie op de Grote Vraag: kunnen we chemische reacties voorspellen door berekeningen, in plaats van ze daadwerkelijk uit te voeren. Dat is namelijk veel goedkoper, omdat computers minder kosten van dieren(levens).
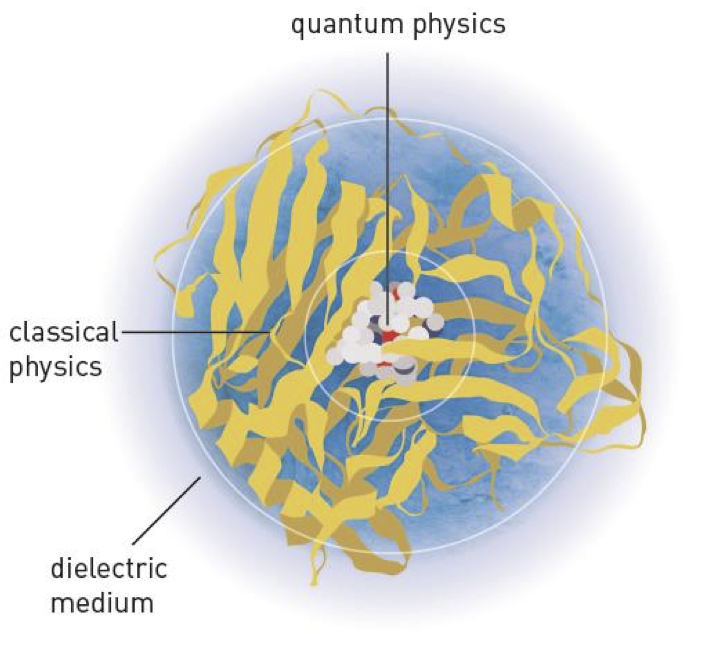
Een medicijn is in de praktijk meestal een molecuul dat hecht aan een plek in het lichaam, bijvoorbeeld een eiwit. Die aanhechting is een chemische interactie. Er zijn twee manieren om een chemische reactie te voorspellen door middel van simulaties. De ene begint met kwantum mechanica, de andere met klassieke mechanica.
Laten we beginnen met de klassieke mechanica. Dit is het soort interactie dat je kent uit het dagelijks leven. Je trapt tegen een voetbal en de voetbal krijgt een impuls. Of: je houdt een magneet bij een stuk ijzer en je voelt de aantrekkingskracht. Er zijn relatief eenvoudige formules om dat soort effecten te uit te rekenen. Als twee eiwitten interactie met elkaar vertonen, zou je dit op de klassieke manier uit kunnen rekenen. Een computational chemist zit dan een tijdje achter z’n computer, download van een speciale website de structuur van de eiwitten, en slaat aan het programmeren. Na een tijdje zal hij met een paar antwoorden komen: zo-en-zo zullen deze twee eiwitten waarschijnlijk interactie met elkaar hebben. Een experimentalist zoals een celbioloog of biochemicus kan dat controleren door in het lab het experiment te doen: doe de twee eiwitten bij elkaar en meet met bijvoorbeeld kristallografie hoe ze in elkaar zitten. Dat klinkt eenvoudig, maar dit soort experimenten kunnen weken of maanden duren. Als dit soort experimenten überhaupt wel lukken.
De andere manier om chemische reacties te voorspellen, gebruikt lessen uit de kwantummechanica. En dat is moeilijk, want die werkt op een hele andere manier dan we in de gewone mensenwereld gewend zijn. Er wordt uitgegaan van individuele atoomkernen die dicht bij elkaar in de buurt zitten. De elektronen die daar omheen zitten ‘koppelen’ de kernen aan elkaar vast, en zo ontstaat een chemische binding. Hoe die elektronen zich gedragen (en waar dus de locatie van de kernen zitten) kan uitgerekend worden. Een theoretisch chemicus tekent in een speciaal computerprogramma een schatting van de structuur van het molecuul, moet daaromheen een heleboel aannames maken, en laat zijn/haar computer dan een hele tijd rekenen. De uitkomst is dan de precieze locatie van de atoomkernen en de verdeling van de elektronen daar omheen. Het voorspellen van reacties gaat op dezelfde manier: er worden nu twee moleculen getekend, de computer berekent wat de elektronen doen en er rolt een gecombineerd molecuul uit. Wederom: mijn uitleg is een versimpeling van de werkelijkheid en er wordt op het moment enorm veel onderzoek gedaan naar alle aannames die de berekeningen moeten helpen. Het probleem is namelijk dat een klein molecuul uitrekenen binnen een afzienbare tijd te doen is, maar hoe groter het molecuul, des te groter en langer de berekening wordt.
En dat is precies het probleem dat bovenstaande heren aanpakken. Zij hebben een slimme combinatie gemaakt van bovenstaande methodes. De kwantummechanische berekening is namelijk erg betrouwbaar maar duurt lang, en de klassieke berekeningen zijn relatief snel, maar zijn minder exact. Stel dat de heren willen weten hoe een klein molecuul aan een veel groter eiwit bindt. In die computerberekening wordt het losse molecuul en de bindingsplaats van het eiwit kwantummechanisch aangepakt, terwijl de rest van het eiwit volgens de klassieke methode wordt berekend. Een ontzettend slimme combinatie van snelheid en nauwkeurigheid. (Nu zeg ik wel “snelheid”, maar zo’n berekening kan nog gerust een paar dagen tot weken duren. Als het niet langer is.)
Nu, waar blijven nou de Nederlanders? Ik had beloofd te vertellen waar die in beeld zouden komen. Het zit zo. In Nederland zijn chemici sinds een lange tijd geïnteresseerd in het uitrekenen van moleculaire processen. In 1976 was er een CECAM-bijeenkomst (Center Europeen Calcul
Atomique et Moleculaire) waar de grondslag voor veel van bovenstaande theoriën werden gelegd. Herman Berendsen en Wilfred van Gunsteren (PDF) (beiden toen in Groningen) hebben toendertijd met Martin Karplus en Michael Levitt tijdens die CECAM-meeting de eerste moleculaire simulaties gedaan. In die tijd waren goede computers zeldzaam (en bestonden toen nog uit ponskaarten), en tijdens de 8-weken durende workshop werd de grondslag voor de toekomst gelegd. Die simulaties hebben geleidt tot de ontwikkeling van drie belangrijke softwareprogramma’s: CHARMM, GROMOS (later Gromacs) en AMBER. Die drie vormen nu de basis van praktisch alle andere softwarepakketten waar moleculaire berekeningen mee gedaan worden.
Kortom: als de Nobelprijs niet aan drie, maar aan zes mensen uitgedeeld, zaten er zeker twee Nederlanders tussen die destijds in Groningen werkten. Desalniettemin vinden wij het onderzoek van de Nobelprijswinnaars van vandaag ontzettend gaaf en we hopen dat dit soort onderzoek nog lang mag duren en vruchtbaar zal zijn.
Update Afgelopen week hebben we hard ons best gedaan om de Nobelprijs voor de Scheikunde in De Wereld Draait Door te krijgen. Ook dit jaar zijn we op het laatste moment afgezegd. Wel hebben we een interview aan het ANP afgegeven, dat is geciteerd door het Reformatorisch Dagblad en NU.nl.
Met dank aan prof. dr. Alexandre Bonvin
Een gedachte over “Nobelprijs Scheikunde naar computerberekeningen aan moleculen”